The Role of Circulating Immune Complexes in Coronary Artery Lesions: Understanding possible Patho-Physiology in Kawasaki Disease Revisited
Author'(s): Saji Philip1*, Ankur Jindal2 and Krishna Kumar R3
1Tiruvalla Medical Mission Hospital, Kerala, Aster Medicity,Kochi, Kerala, India.
2Pediatric Clinical Immunology & Rheumatology, Post Graduate Institute of Medical Education, Chandigarh, Punjab, India.
3Department of Pediatric Cardiology, Amrita Institute of Medical Sciences & Research Centre, Kochi, Kerala, India.
*Correspondence:
Dr. Saji Philip, MB,BS, DCH, F. Card (APSSEAR), Ph.D, FAHA. Fetal & Pediatric Cardiology Services, Founder President of Indian Society of Kawasaki Disease, Tiruvalla Medical Mission Hospital, Kerala, India 689101. Email; tfcsaji@yahoo.co.in.
Received: 02 Nov 2022; Accepted: 03 Dec 2022; Published: 08 Dec 2022
Citation: Philip S, Jindal A, Krishna Kumar R. The Role of Circulating Immune Complexes in Coronary Artery Lesions: Understanding possible Patho-Physiology in Kawasaki Disease Revisited. Cardiol Vasc Res. 2022; 6(5): 1-12.
Abstract
Kawasaki disease (KD) is an acute self-limiting systemic vasculitis of unknown etiology affecting predominantly the coronary arteries. The role of circulating immune complexes (ICs) in the pathogenesis of KD has been studied using the sera of patients with KD. It has been proposed that ICs are triggered by single or multiple unknown causative agents as well as vasculitis. An outbreak of SARS CoV2 infection caused similar pathophysiology in producing vasculitis, and the RNA virus may have triggered signs and symptoms similar to KD. For clinicians and researchers alike, detecting the causative agents of KD remains a challenge. According to studies in animal models, type III hypersensitivity reactions caused by serum sickness are a prototype for immune complex vasculitis. The signs and symptoms of coronary artery dilation in swine are similar to those of KD. These models may be used to evaluate new pharmacological agents for KD. The pathogenesis of KD is complex and remains inadequately understood at present. However, circulating immune complexes may play a key role in the pathophysiology of KD and coronary artery vasculitis. Various therapeutic agents are being explored in the management of KD and these agents act at various stages of the production of pro-inflammatory cytokines and chemokines. In this review, we discuss recent developments on the pathogenesis of KD and provide insights into the innate immune response and mechanisms behind coronary artery damage in KD. We specifically explore the potential role of ICs in the pathogenesis of KD.
Keywords
Introduction
It has been 55-years since Dr. Tomisaku Kawasaki published his report entitled “mucocutaneous lymph node syndrome” in 1967, now known as Kawasaki disease (KD) [1]. KD is a self-limiting systemic inflammatory disease of childhood with predilection to affect coronary arteries. The etiology of KD, remains an enigma for researchers. KD is the most common cause of acquired heart disease in the young in developed countries [1-3]. During the recent pandemic, KD like illness was reported in patients who suffered from COVID-19 infection. The pandemic has indicated that an infectious agent may play a key role in triggering KD, predominantly by the mechanism of immune complexes (ICs) formation [4]. Although the etiology of KD remains unknown, a double hit hypothesis involving an infectious trigger in a genetically predisposed individual has been proposed [5,6]. The clinical and epidemiological features of KD strongly suggest an infectious etiology, including the occurrence of epidemics, seasonal variation in incidence and clustering of cases [7]. Unlike most infections, however, there are significant differences in racial predilection that suggest a strong genetic influence. There is possibility of triggering ICs by various infectious and non-infectious agents in the pathogenesis of KD. In the present review, we update on various studies that have shown the role of circulating ICs in the pathogenesis of KD.
Circulating Immune Complex
Antigen-antibody complexes are formed when antibodies are produced against a circulating or tissue antigen. The antigen may be exogenous from an infectious agent, toxin or drug, or may be endogenous as occurs in autoimmune disorders. ICs are formed during many infectious and inflammatory diseases, and may play an important role in immunopathogenesis of infectious and inflammatory processes. They are normally taken up by inflammatory cells through binding of the heavy chain constant region to immunoglobulin Fc receptors (FcRs). Binding of immunoglobulin to some classes of FcRs (FcgRI, FcgRIIA, FcgRIIC, FcgRIIIA, FcgRIIIB) may lead to activation of inflammatory cells while binding to FcgRIIB result in suppression of inflammation. ICs may bind to FcRs and activate several inflammatory cells including monocytes, basophils, eosinophils, lymphocytes and neutrophils [8]. Circulating immune complex may explain immuno-pathogenic mechanisms that lead to symptoms and signs of vasculitis in KD [9-13].
Physiological and Pathological Aspects of Circulating Immune Complexes
Immune complexes (ICs), production, due to interaction of foreign substances with specific antibodies, constitutes an essential part of normal human immune defence mechanisms. This reaction is generally followed by one or more secondary reactions, all of which enable the body to neutralize and clear microorganisms and non- self-molecules (in the form of ICs after antibody binding) that have penetrated the various body barriers. Inactivation and elimination of these "invaders" prevents their deposition (localization) where they might multiply (in the case of microorganisms) or induce specific damage (toxins or enzymes) [14]. ICs formation followed by these secondary reactions (such as complement fixation) enhances macrophage system clearance (MPS) mechanisms and prevents interaction with specific sites in the body that could be damaged by deposition. ICs do not normally accumulate in blood or organs, however, there are circumstances under which potentially pathogenic ICs might form in the circulation and not be cleared properly. Factors that could influence this phenomenon and the manifestations of specific disease activity include the nature and quantity of the antigen and the antibody response, and the state of the systems involved in ICs clearance (complement, complement receptors, and receptors on fixed cells of the MPS for both complement components and the Fc region of immunoglobulins) [14].
There are an infinite number of potential antigens (from whole organisms to small peptides), and the antibody response may vary with respect to class, subclass, affinity, etc. As a result, characteristics of ICs would be quite variable and analysis of one ICs system may not be applicable to other systems as well. A few examples are as follows: A patient with a monoclonal antibody against flavin became "yellow" because of the ubiquitous accumulation of flavin. Apparently, the antibody/flavin immune complex was cleared from the circulation at a much slower rate than flavin alone; the small ICs (one antibody per ICs) was too small to be cleared by the Mononuclear Phagocyte System (MPS), yet clearance of the flavin was apparently blocked by the antibody. Presence autoantibodies to amylase, prostatic acid phosphatase or creatine kinase can block the clearance of these enzymes by a similar mechanism [15]. Nephritic factor is an autoantibody that stabilizes alternative pathway C3 convertase in the circulation, thus inducing C3 depletion. These examples illustrate one end of the spectrum wherein antigen circulates for prolonged period of time because of it complexes with specific antibody and forms a small ICs [13].
At the opposite end of the spectrum are patients with mixed essential cryoglobulinemia. Large quantities of precipitating ICs (containing IgG and/or 1gM, and specific antigens) deposit at sites throughout the body, including the glomeruli [16]. These ICs can fix complement and cause local damage at their sites of deposition. It is likely that many ICs that form in the circulation will have properties intermediate between these two extremes. The balance between rapid and safe clearance versus tissue localization will then be influenced by factors that have been described in part in experimental studies but have yet to be well defined in humans. These include the potential affinity of antibody or antigen for specific tissue (such as, DNA for glomerular basement membrane), and the hemodynamic and inflammatory status of the individual [17].
The specific immunochemical properties of the ICs, and in particular their potential to interact with Fc receptors and to fix complement, and react with complement receptors will fundamentally influence their ultimate fate and rate of clearance from the circulation [13,18]. ICs formation and clearance cannot be a steady state process; even during chronic serum sickness the concentrations of antigen, antibody, and ICs formation may vary continuously [13,16,17]. Thus, ICs could form and deposit in a brief period. For example, patients with essential mixed cryoglobulinemia have sudden episodes of purpura over their legs and arms, and then their vasculitis subsides despite the presence of measurable ICs [16].
Circulating Immune Complexes in Patients with Kawasaki Disease, Triggered by Infectious Agents
Circulating ICs, triggered by infectious agents such as, bacteria, virus or other unknown agents, have been detected in the early phase of patients with KD and, might be involved in the immunopathological mechanisms of development of vasculitis in these patients [7]. Although many pathogens have been implicated via superantigen toxins, staphylococcal and streptococcal toxic shock syndromes, other studies have failed to confirm these findings [19-22]. Several bacterial such as Yersinia pseudotuberculosis [23-25], Propionibacterium acnes [26], Mycoplasma pneumonia [27,28], Chlamydia pneumonia [29], Rick ettsia species [30] and Coxiella burnetii [31], pseudomonas [32] and several viral agents such as Epstein Barr virus [33], retroviruses [34,35], adenovirus [36], measles virus [37]. In addition, fungal agents such as candida [38] have been reported to be associated with KD. The novel coronavirus responsible for Severe Acute Respiratory Syndrome

Coronavirus 2 (SARS-COV-2), is known to induce a systemic inflammatory response affecting multiple organs, with lungs being most common and severely affected. Some of the extrapulmonary manifestations include involvement of the systemic vasculature, similar to KD. Several case reports have shown that SARS-COV-2 stimulates an immune reaction mimicking KD [39]. Singh et al., reported association of KD with influenza [40]. Nakamura et al. reported that pattern of KD got affected in 2009 after the epidemic of influenza A/h3N1 in Japan and observed a documented source of infection, both bacterial and viral in one third of patients with typical KD [41]. Furthermore, Rowley et al. showed presence of cytoplasmic inclusion bodies that are compatible with the viral protein aggregations and nucleic acid in ciliated bronchial epithelium of patients with KD in the acute phase [42]. Considering the infectious cause for KD, a case-control study by Esper et al. suggested an association between human coronavirus- New Haven (HCoV-NH) infection and KD [43]. None of these reported viruses or bacteria have been convincingly replicated. A summary of studies on circulating immune complexes in KD patients by different methods of detection showed in Table 1, suggestive of definite triggering agents behind this CIC causing vasculitis.
Horse Serum Induced Type III Hypersensitivity Reaction
An attempt was made to produce immune complex vasculitis in swine, hoping to produce as an ideal experimental coronary artery disease (CAD) animal model by administrating foreign protein, such as horse serum [44,45]. Philip S et al. had studied 21 pure bred male piglets of 1.5, 2, 3 months of age with normal saline and HS and the results of clinical observations, hematoserology, echocardiography, histopathology were very much similar to patients with KD. 2-D echocardiogram of coronary arteries of piglets who received horse serum showed coronary intimal irregularities and aneurysmal dilatation. (Figures 1. A, B, D, E). Pericardial thickening as evidence of pericarditis was also observed (Figure 1C). Type III hypersensitive reaction is induced by antigen-antibody complexes that produced tissue damages as a result of their capacity to activate a variety of serum mediators principally the complement system. The localization of circulating immune complex in experimental serum sickness and correlation of possible immune complex mechanism in KD was already studied by Knicker et al. [46], Fossard C et al. [47], and Cochrane et al. [48] Hence clinical and histopathological findings of systemic type III hypersensitivity reaction may mimic KD and possible similar mechanism may be involved in the pathogenesis of coronary arteritis in KD [44,45] (Figure 1F & Figures 2D&C). Possible mechanism of damaging vascular endothelial cells by immune complexes in composite of drawing showed in Figure 2. A-B & Figure 3. A-H).
The pathology of KD has been studied in details and immune complexes may play an important role in the pathogenesis of its vasculitis in swine [47-68]. Histopathology results after 14-60 days of HS infusion showed subacute to chronic phase of arteritis such as intimal proliferation, necrosis, vacuolization, smooth muscle cells proliferation etc (Figure 1F & Figure 3 A-H).
Histopathology of specimens of HS group revealed peri arteritis and pan-venulitis in the acute phase including cellular infiltrations

Figure 1 A-F: A,B,D,E; 2-D echo cardiogram showed dilatation and intimal irregularities of proximal LAD in piglet received HS at 4th day. C; showed pericardial thickening and F; H&E staining of proximal LAD showed intimal and inner third of intima-medial proliferation with edematous changes at 14-days. HS; horse serum, LAD: left anterior descending artery, H&E; Hematoxylin and eosin.
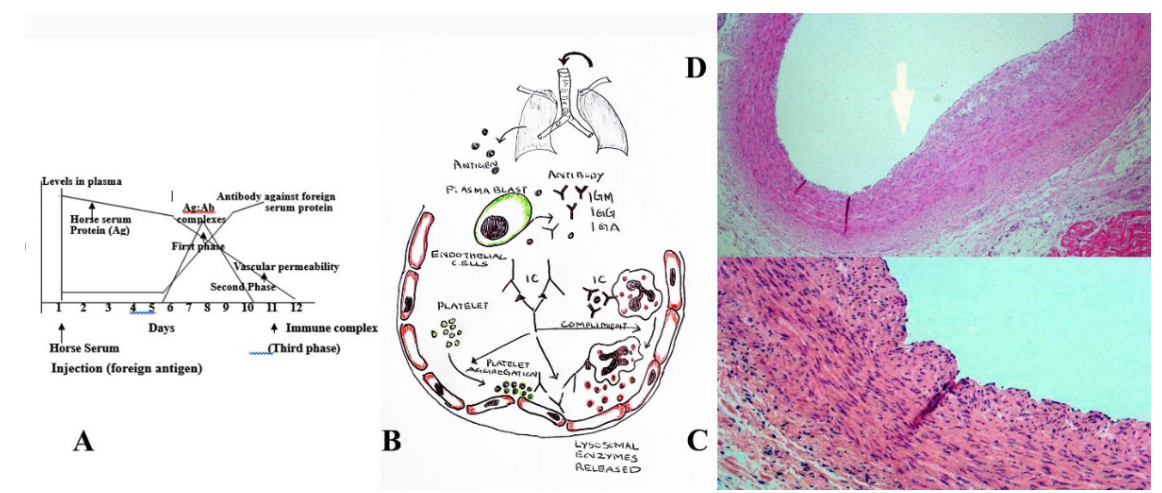
Figure 2 A-D: Induction of immune complex vessel wall injury. Figure 1 A; Three sequential phases in induction of systemic type III sensitive reaction. B; Composite of drawing depicts mechanism of immune complex vessel wall injury. Antigen possibly enters through the respiratory tract as an unknown infectious or noninfectious agents producing specific antibodies from plasmablast. Further formation of Immune complex leads to cascade of platelet aggregation, releasing of lysosomal enzymes from the neutrophils, compliment fixation and damages the endothelium. C&D; showing how histopathological changes occurring of proximal left anterior descending artery showed (hematoxylin and eosin stain) intimal proliferation (C) and intimal and inner third medial proliferation induced by horse serum mediated immune complex coronary changes.
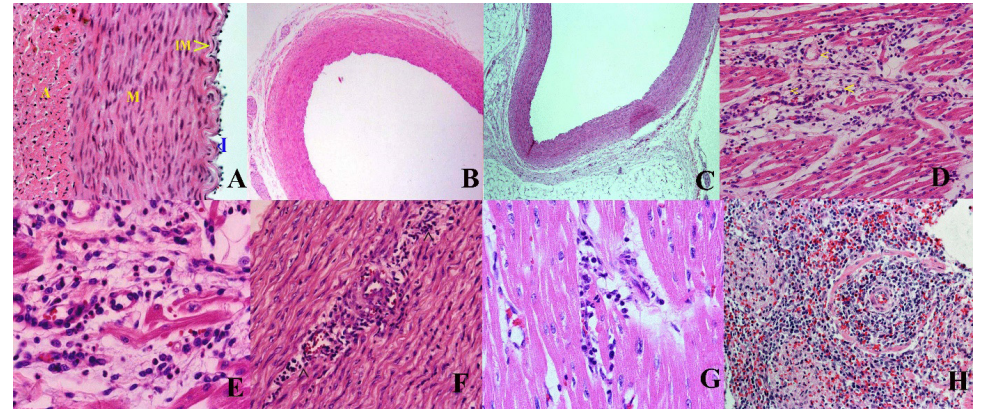
Figure 3 A-H: Histopathology of coronary arteries (H&E staining) of horse serum induced coronary vasculitis in the saline and HS groups of swine. (A-C) showing normal coronary arterial walls after three doses of normal saline infusions in the saline group. (A) Left coronary artery (400x) at Day 41 showing normal intima (I), internal elastic membrane (IM), tunica media (M), and adventitia (A). (B) Left coronary artery (40x) at 40 days (C) Left anterior descending artery (40x) at Day 24 showed normal. D-H; H&E staining of coronary arterial walls of piglets in the HS group. (D, E; 2-4 days) Perivenular cellular infiltrates of the coronary vein and vasa vasorum (<) (200x) and diffuse cellular infiltrates in the tunica media (400x) at Day 3. (F) Cellular infiltrates in the tunica media (200x) of the ascending aorta at Day 2. (G) Diffuse cellular infiltrates in the myocardium (400x) and (H) in the distal tubular areas of the right kidney (200x) at Day 2. HS; Horse serum, H&E; Hematoxylin and eosin within myocardium (Figure 3D-F). Autopsy after 28 hours showed more infiltrations than 48 hours specimen (Figure 3F). Whereas saline group had normal coronaries (Figure 3A-C). Immune complex vascultis changes observed in HS infusion group may serve as an ideal experimental animal model for coronary vasculitis mimicking KD, especially testing the efficacy of pharmacological agents in prevention of coronary artery aneurysms. Albumin that acts as an exogenous foreign protein antigen in HS group produces antigen antibody complexes leading to systemic hypersensitivity reaction. Onouchi et al. also reported that horse serum induced immune complex vasculitis in rabbit was very much similar to the pathophysiology of CAD in KD [69].
Kawasaki Disease and Sars-Cov2 Infections
Systemic inflammation is the most striking finding in some patients during COVID-19 infection. SARS-CoV2 is a viral disease with inflammation and infection of endothelial cells. Presence of viral elements and inflammatory cells within endothelial cells and the evidence of endothelial cell death suggesting that SARS-CoV-2 infection facilitates endothelium cell inflammation through direct viral involvement and inflammatory response [70]. SARS- CoV2 induced endothelium cell inflammation may explain systemic microcirculatory dysfunction in different vascular beds. Moreover, it has been suggested that the induction of apoptosis and pyroptosis (represents a form of cell death that is triggered by proinflammatory signals and associated with inflammation) might have an important role in endothelial cell damage in patients with SARS-CoV2. The functional receptor for the entrance of the SARS-COV-2 into the cytoplasm is angiotensin convertase enzyme (ACE) inhibitor [71]. The classical axis of the renin-angiotensin- aldosterone system (RAS) consists of ACE, angiotensin II (Ang II), and angiotensin receptor type 1 (AT1). This system could induce tissue injury, inflammation, and fibrosis. In contrast, ACE2 employs the opposite effects on tissue fibrosis and inflammation by converting Angiotensin II, which has anti-inflammatory, anti- proliferative, and anti-fibrotic properties. It appears that children have shown a less severe form of COVID-19 likely due to the high ACE2 receptor concentrations, as well as a more qualified immunity, and a constitutional high lymphocyte rather adults.
On the other hand, the ACE2 gene maps to chromosome Xp22, exhibits a high degree of genetic polymorphism [72]. It has been reported that SARS-CoV2 down-regulates ACE2 receptor is carried out through the spike protein of the virus (SARS-S) via a process that is tightly coupled with TNF-α production. It may be hypothesized that KD-like disease in children following SARS- CoV-2 might be because of influence of immunomodulatory function of ACE2 and Angiotensin and these patients might be having a lower expression of ACE2 receptors as compared to children who do not develop KD-like disease following SARS- CoV-2 infection. Genetically under-expression of ACE2 receptor in children with genetically-susceptible to KD who are infected with SARS-CoV-2 possibly further downregulates the ACE2 expression by TNF-α and leads to surge of inflammation including TNF-α and progression to Kawasaki-like disease [73]. Once the criteria for KD is full filled, you may treat as KD and we know that triggering agent is RNA virus in SARS Co-V2 infection.
The profile of the cytokine storm associated with severe COVID-19 disease is similar to that of secondary hemophagocytic Lympho-histiocytosis (HLH), which is a rare complication of other viral infections (3.7-4.3%). Secondary HLH is characterized by fulminant and fatal hyper-cytokinemia with multiorgan failure. In severe infection, lower peripheral lymphocyte counts (CD4 and CD8 T cells), higher interleukin (IL) levels (IL-6 and IL- 10), decreased interferon-gamma expression in CD4+ T cells and higher D-dimer and fibrin degradation products (FDP) levels, leading to increased thrombosis and multiorgan injury has been described [72]. Moreover, patients with severe infection may also have abnormal coagulation parameters, perhaps related to high expression of ACE2 receptors in vascular endothelial cells. Antioxidant trial in HS induced vasculitis was quite successful in swine model [74]. In addition, unpublished data of antioxidant trial such as Vitamin E, A and C trial in patients with KD was found to be quite successful in mitigating vasculitis in both signs and symptoms. Hence antioxidants also may be helpful in mitigating vasculitis along with intra venous immunoglobulin with or without methyl prednisolone.
Hyper Inflammation and Cytokine Storm
Besides the direct cytopathic effect of the virus, the host's immune response and lung inflammation play an important role in the pathogenesis of COVID-19 and may be implicated in disease severity and mortality, making this a potential target for treatment [5]. Increased acute-phase reactants, cytopenias (thrombocytopenia and lymphopenia), coagulopathy (elevated D-dimer), hepatitis (elevated LDH, AST, ALT), and macrophage activation (elevated ferritin) correlate with severity and mortality. There are several hypotheses on how the virus might induce inflammation. Pyroptosis, related to viral infection and replication in airway epithelial cells, leads to cytokine release and consequent vascular leakage. As expected, IL-1β, released during pyroptosis, results in elevated cytokine during SARS-CoV-2 infection. Viral infection of monocytes and macrophages can also result in aberrant cytokine production. These proinflammatory cytokines and chemokines, including IL-6, IFNγ, MCP1, and IP-10, attract immune cells, notably monocytes and T lymphocytes, but not neutrophils. SARS-CoV-2 patients also exhibited high levels of IFNγ and IL-18, which are key players in the cytokine storm syndrome. Elevated levels of cytokines, such as TNF-α, can cause septic shock and MOF. Hyperinflammation is similar to, but not fully overlapping with other well-known clinical entities, such as macrophage-activated syndrome (MAS), or hemophagocytic lympho histiocytosis (HLH) and other forms of viral-induced cytokine storm, in that ferritin increase is modest and severe end- organ disease is limited to the lung. Nonetheless, it is becoming more and more evident that it has a central role in disease severity and outcome. Experience from hyperinflammation in HLH, MAS, and cytokine release syndrome suggests that early intervention is essential to avoid irreversible tissue damage [71].
Pathogenesis and Complications in KD
Most cases of KD are self-limiting and have an uncomplicated course. Although the etiology of KD remains unknown, a double hit hypothesis involving an infectious trigger in a genetically predisposed individual is proposed [8]. Many viruses have been isolated from patients with KD, including adenovirus, Epstein- Barr virus (EBV), human immunodeficiency virus (HIV) and parvoviruses. Viruses are known to trigger a cascade of immune pathways in patients, such as the activation of cyclic guanosine monophosphate-adenosine monophosphate (cGAMP) synthase (cGAS) pathway. cGAMP is one of the deoxyribonucleic acid (DNA) sensors in the body that helps to identify foreign viral DNA material within cells. Once foreign DNA is detected, cGAS produces cGAMP that triggers the activation of simulator of interferon genes (STING) within the endoplasmic reticulum, resulting in the release of cytokines and inflammatory molecules such as type 1 interferons (IFN) as part of the immune response towards the foreign infective agent. STING also acts on the retinoic acid inducible gene-1 (RIG-1), a ribonucleic acid (RNA) sensor and mitochondrial antiviral signalling protein (MAVS) to detect viral RNA and trigger an immune response, indicating a role of the STING pathway in protecting against RNA viruses as well. The STING pathway has been found to be activated in KD and increased IFNs, neutrophils and cytotoxic T cells have been reported in the histology of coronary artery tissue from patients with KD.
Inflammatory markers such as interleukin (IL) -6 and vascular endothelial growth factors (VEGFs) are released by cells of the innate immune system such as monocytes, macrophages, and dendritic cells leading to inflammation of vessels. It has been suggested that an autoantigen located in the walls of coronary arteries may serve as a target for these inflammatory cells and cytokines and leads development of coronary artery aneurysms in patients with KD [74]. However, this hypothesis is yet be proven. Apart from inducing inflammation, neutrophils, macrophages and dendritic cells have also been found to invade into the artery walls, causing damage because of their cytotoxic activity. There also appears to be an excessive activation of cytotoxic CD +8 T cells, further supporting the possibility of an infectious agent as a trigger for the immune activation seen in KD [5]. Immune complexes have been reported in plasma and serum of patients with KD since 1977. They are seen within the first week of illness gradually increase in number and then wane of in 3- 4 weeks. This may suggest that immune complexes are formed in response to a foreign antigen to enhance the targeted action of other inflammatory cells and stimulate phagocytosis of the antigen to remove it from the circulation. While this is an interesting finding, there has been no significant correlation between immune complexes and the severity of illness in KD patients, likely due to incomplete data collection and difficulties in identifying the phases of illness during which test samples were collected. Studies that are more recent are needed to investigate the role of immune complexes in the pathogenesis of KD [75]. An exorbitant immune system activation may occur in KD causing macrophage activation syndrome (MAS), a rare complication that can occur at any stage of KD and lead to increased cardiac complications and high mortality, highlighting the importance of timely diagnosis and management [76].
Studies have shown that this immune response is strongest during the first few weeks of infection with many inflammatory molecules such as cytokines and C-reactive protein being released into the bloodstream. This could be related to the increased expression of cytokines such as IL-1, IL-6 and IL-8 [75]. Tumor necrosis factor- alpha (TNF-α) levels are raised more significantly in patients with KD who develop coronary artery aneurysms as compared to those without. This could be related to TNF-α triggering the release of chemokines by endothelial cells, increasing permeability of the endothelial lining to other inflammatory cells such as neutrophils and causing more severe damage to the affected arteries. Superantigen binds to T cell receptor induce release of immune mediators IL6, TNF alpha, TGF beta. Identification of CD8+, T lymphocyte, IgA plasma cells and macrophages in coronary arteries suggest viral etiology [77]. Hence, both infections and immune component actively takes part in the pathogenesis of KD (Figure 3B).
Circulating Platelet-Neutrophil Aggregates play a significant role in KD
Platelet activation at the site of inflamed endothelium contributes to vascular inflammation and vascular wall remodelling. Released chemokines from activated platelets, such as platelet factor 4 (PF4), CXCL7 and β-thromboglobulin (β-TG), have important effects on vascular inflammation. Vascular injury may lead to increased platelet activation with neutrophil and monocyte infiltration, as well as increased platelet adhesion and aggregation via the release of inflammatory cytokines. Circulating platelet- neutrophil aggregates amplify acute inflammation and exhibit a hyper-reactive response that could promote the development of thrombotic and inflammatory disease, obstruct the flow of coronary micro vessels, and contribute to vascular inflammation and tissue injury. Likewise, activated platelets and neutrophils have been demonstrated during the acute phase of KD-associated inflammation and may be contribute to the occurrence of coronary artery aneurysm [78]. Therapeutic inhibition of platelet-neutrophil aggregates reduces neutrophil recruitment and permeability and may help to attenuate organ damage and mitigate the inflammatory process. Cortico steroids may cause inhibition of platelet adhesion, spreading, aggregation, thrombus formation and the interaction of platelets with monocytes through regulation of P2Y12 receptor signalling, which is the main platelet receptor responsible for ADP induced platelet activation. Thus, prednisolone has a role in helping to control vascular and thrombotic Diseases. The use of corticosteroids as part of a combination treatment may have a beneficial effect on terminating the inflammatory process by its anti-inflammatory property, which suppresses immune cell activation, proliferation, and cytokine production, as well as by its ability to decrease endothelial expression of cellular adhesion molecules in the acute phase of KD. However, previous studies have suggested that the use of corticosteroids for patients with KD should be limited because such treatment may be linked to a higher incidence of CAA and impaired vascular remodelling. The main benefit of corticosteroid combination treatment is considered early suppression of the vasculitis that precedes vascular remodelling [78].
Pathways for Nuclear factor Kappa B (NF-κB) Signalling in the Cytoplasm and the Mitochondrion
Macrophages play key role in the inflammatory process. Angiotensin II in turn release NADPH (Nicotinamide adenine dinucleotide phosphate is a required cofactor for CYP-mediated biotransformation, and oxygen serves as a substrate). Singlet Oxygen releases from the NADPH oxidase and singlet oxygen convert low-density lipoprotein to oxidised LDL that in turn release the transcription factor nuclear factor kappa B (NF-κB), regulates multiple aspects of innate and adaptive immune functions, and serves as a pivotal mediator of inflammatory responses (Figures 4 & 5).
NF-κB tri-subunit complex exists in an inactive state in the cytoplasm. NF-κB activation is initiated by TNF-α binding to TNF receptors. Intrinsic apoptotic pathway stimulation by NF- κB activation in mitochondria leads to cytochrome c release, thus triggering caspase cascades and programmed cell death. NF-κB induces the expression of various pro-inflammatory genes, including those encoding cytokines and chemokines, and participates in inflammasome regulation. In addition, NF- κB plays a critical role in regulating the survival, activation and differentiation of innate immune cells and inflammatory T cells (Figure 5). Consequently, deregulated NF-κB activation contributes to the pathogenic processes of various inflammatory diseases [79,80].
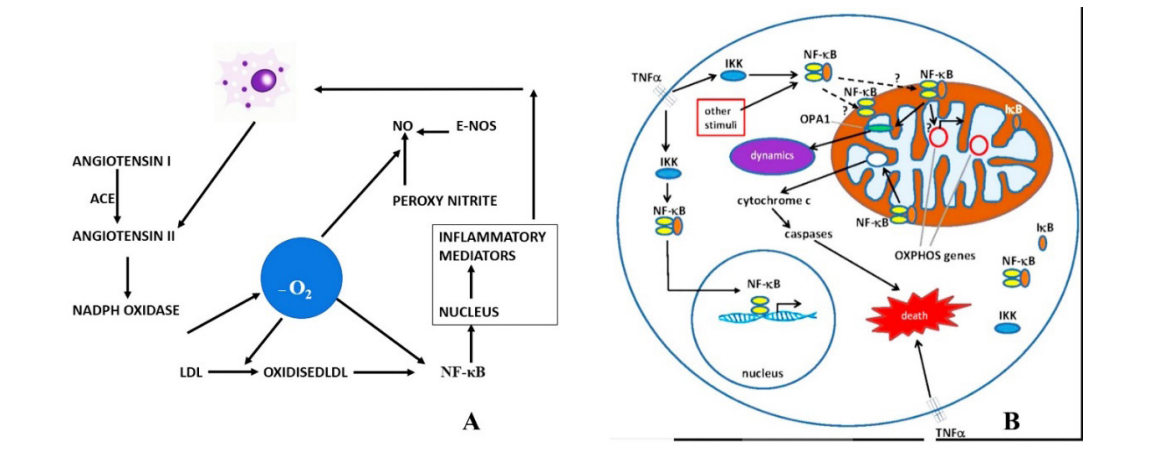
Figure 4 A-B: A; Shows role of macrophages and various process in turns release of singlet oxygen leading to inflammatory mediators through NF-κB. B; Role of TNF alpha in the production of NF-κB at the mitochondrial level leading to inflammatory cascade with cellular death. NADPH; Nicotinamide adenine dinucleotide phosphate, NF-κB transcription factor nuclear factor kappa B. e-Nos; endothelial nitric oxide. ACE: angiotensin converting enzyme. AT II; angiotensin II, NO; Nitric oxide
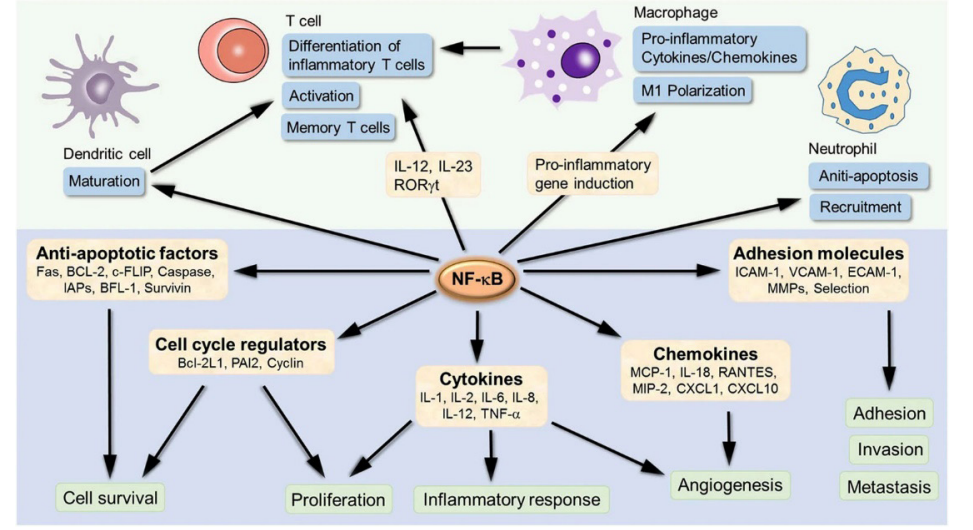
Figure 5: NF-κB induces the expression of various pro-inflammatory genes, including those encoding cytokines and chemokines, and participates in inflammasome regulation. NF-κB is an inducible transcription factor, which can activate transcription of various genes and thereby regulate inflammation. NF-κB target inflammation not only directly by increasing the production of inflammatory cytokines, chemokines and adhesion molecules, but also regulating the cell proliferation, apoptosis, morphogenesis and differentiation.
Role of Antioxidants in Mitigation of Vasculitis
The goal of initial management in KD is to reduce inflammation. Intravenous gamma globulin and aspirin form the gold standard regime. A multicentre randomized controlled trial in the United States demonstrated the effectiveness of the combination in lowering coronary artery lesions in KD. Antioxidants can reverse endothelial dysfunction induced by methionine and restore the endothelial function in hyperlipidaemia, and they can slow down the thickening of arteries. A trial of antioxidants in prevention of coronary artery leisions in horese mediated vasculitis in swine model showed potential relevance of antioxidants as an add on therapy in mitigation of coronary vasculitis in KD [74]. The role of antioxidants in protection against cardiovascular disease prevents endothelial dysfunction in humans by regulating endothelial NO levels as well as by inhibiting cardiovascular inflammation, lipid peroxidation, platelet aggregation, and low-density lipoprotein oxidation [74].
Reactive oxygen species generated by activated polymorpho- neutrophils are directly toxic to endothelial cells and are a potent cause of endothelial cell damage. The role of antioxidants in protection against cardiovascular disease prevents endothelial dysfunction in humans by regulating endothelial NO levels as well as by inhibiting cardiovascular inflammation, lipid peroxidation, platelet aggregation, and low-density lipoprotein oxidation [81]. Chain-breaking antioxidants such as vitamins C and E are powerful reductants that scavenge free radical species to prevent further oxidation. Phagocytic cells, including neutrophils and macrophages, generate NO via NO synthase that is inducible by immunological stimuli such as endotoxin (Lipopolysaccharides) and various cytokines [82]. The broader role of NO in the inflammatory response is not well established, although the reactivity of NO or its potential conversion product, peroxynitrite anion, with sulfhydryl groups indicates the possibility of cellular biochemical targets, the alteration of which would put tissue at risk. The protective effects of NO synthase in immune complex- induced vasculitis were also studied by Mulligan et al. [83]. There is increasing evidence that lipoprotein oxidation plays an important role in the vascular endothelial damage and atherosclerosis [83]. In various areas of this cycle can break and can mitigate the pro-cytokines and further inflammations in KD coronary artery lesions (Table 2). LDL to oxidised LDL and can be reduced by administration of vitamin C, E, Statin and PPAR (PPAR agonists are drugs which act upon the peroxisome proliferator-activated receptor. They are used for the treatment of symptoms of the metabolic syndrome, mainly for lowering triglycerides and blood sugar). Same way NFkB and NADPH oxidase can be reduced by giving statin and PPAR agonist. Vitamin C and Statin have role in reducing release of singlet oxygen from endothelial nitric oxide (Figure 4). TNF alpha and NFkB inhibitors can stop apotosis or celluar damages in the coronary endothelium (Figure 5).

Conclusions
The specific etiology remains inadequately understood in Kawasaki disease. However, circulating immune complexes triggered by various etiologic agents play a key role in the pathogenesis of coronary vasculitis in KD. Horse serum-induced Type III hypersensitivity reaction in swine indicates that serum sickness is a prototype of immune complex vasculitis, and the signs and symptoms of coronary artery dilation in swine resemble those of KD. Therefore, interrupting immune complex formation via treatment trials either in isolation or in combination, targeting various steps in the pathogenesis of coronary artery lesions, such as the production of inflammatory cytokines, chemokines, TNF alpha, NO, bacterial superantigens and specific pathogenic autoantibodies, will certainly be given a ray of hope.
Acknowledgement
Prof. Ming-Tai, Department of Pediatrics, Division of Pediatric Cardiology, National Taiwan University Hospital, Taipei, Tawan (for active discussions on role of Immune complexes in KD).
References
- Kawasaki T. Acute febrile mucocutaneous syndrome with lymphoid involvement with specific desquamation of the fingers and toes in children: clinical observations of 50 cases. Arerugi (Jpn J Allergol). 1967; 16: 178-222.
- Burns JC, Glodé MP. Kawasaki syndrome. Lancet. 2004; 364: 533-544.
- Newburger JW, Takahashi M, Burns JC. Kawasaki Disease. J Am Coll Cardiol. 2016; 67: 1738-1749.
- Menikou S, Langford PR, Levin M. Kawasaki Disease: Role of Immune Complexes Revisted. Front Immunol. 2019: 10: 1156-1166.
- Fang Y, Aravamudan VM, Sridharan GK, et al. Kawasaki like illness due to COVID-19: a review of the literature. J Infect Dev Ctries. 2021; 15: 630-638.
- Fossard C, Thompson RA. Mucocutaneous lymph-node syndrome ( Kawasaki disease): probable soluble complex disorder BMJ. 1977; 1: 883-884.
- Makino N, Nakamura Y, Yashiro M, et al. Epidemiological observations of Kawasaki disease in Japan, 2013-2014. Pediatr Int. 2018; 60: 581-587.
- Pachman LM, Herold BC, Davis AT, et al. Immune complexes in Kawasaki syndrome: a review. Prog Clin Biol Res. 1987; 250: 193-207.
- Weindling AM, Levinsky RJ, Marshall WC. Hood J 1979 Circulating immune complexes in mucocutaneous lymph- node syndrome (Kawasaki disease). Arch Dis Child. 1979; 54: 241-242.
- Furuse A, Matsuda I. Circulating immune complex in the mucocutaneous lymph node syndrome. Eur J Pediatr. 1983; 141: 50-51.
- Mason WH, Jordan SC, Sakai R, et al. Circulating immune complexes in Kawasaki syndrome. Pediatr Infect. 1985; 4: 48-51.
- Li CR, Yang XQ, Shen J, et al. Immunoglobulin G subclasses in serum and circulating immune complexes in patients with Kawasaki syndrome. Pediatr Infect Dis. 1990; 9: 544-547.
- Shirai N, Yabana T, Ueda M. Pathology of Kawasaki disease. Nippon Rinsho. 2008; 66: 251-257.
- Schifferli JA, Taylor RP. Physiological and pathological aspects of circulating immune Complexes. Kidney International. 1989; 35: 993-1003.
- Fari-Iangi M, Osserman EF. Myeloma with xanthoderma due to an IgG lambda monoclonal anti-flavin antibody. N EngI J Med. 1976; 294: 177-183.
- Ponticelli C, D'amico G. Essential mixed cryoglobulinemia, in Diseases of the Kidney (4th ed) edited by Schrier RW, Gottschalk CW, Boston, Little Brown & Co. 1988; 78: 2377-2392.
- Couser WG. Mechanisms of glomerular injury in immune- complex disease. Kidney mt. 1985; 28: 569-583.
- Couser WG, Baker PJ, Adler. Complement and the direct mediation of immune glomerular injury: A new perspective. Kidney Int. 1985; 28: 879-890.
- Xu SX, McCormick JK. Staphylococcal superantigens in colonization and disease. Front Cell Infect Microbiol. 2012; 2: 52.
- Curtis N, Chan B, Levin M. Toxic shock syndrome toxin- secreting Staphylococcus aureus in Kawasaki syndrome. Lancet. 1994; 343: 299.
- Morita A, Imada Y, Igarashi H, et al. Serologic evidence that streptococcal superantigens are not involved in the pathogenesis of Kawasaki disease. Microbiol Immunol. 1997; 41: 895-900.
- Gupta-Malhotra M, Viteri-Jackson A, Thomas W, et al. Antibodies to highly conserved peptide sequence of staphylococcal and streptococcal superantigens in Kawasaki disease. Exp Mol Pathol. 2004; 76: 117-121.
- Tahara M, Baba K, Waki K, et al. Analysis of Kawasaki disease showing elevated antibody titres of Yersinia pseudotuberculosis. Acta Paediatr. 2006; 95: 1661-1664.
- Usui D, Ishii Y, Akaike H, et al. Yersinia pseudotuberculosis type 4a infection meeting the diagnostic criteria for Kawasaki disease complicated by disseminated intravascular coagulation. Kansenshogaku zasshi J Jap Assoc Infect Dis. 2005; 79: 895-899.
- Chou CT, Chang JS, Ooi SE, et al. Serumanti- Yersinia antibody in Chinese patients with Kawasaki disease. Arch Med Res. 2005; 36: 14-18.
- Tomita S, Kato H, Fujimoto T, et al. Cytopathogenic protein in filtrates from cultures of Propionibacterium acnes isolated from patients with Kawasaki disease. Br Med J. 1987; 295: 1229-1232.
- Ebrahim M, Gabay M, Rivas-Chacon RF. Evidence of acute mycoplasma infection in a patient with incomplete and atypical kawasaki disease: a case report. Case Rep Med. 2011; 606920.
- Lee MN, Cha JH, Ahn HM, et al. Mycoplasma pneumoniae infection in patients with Kawasaki disease. Kor J Pediatr. 2011; 54: 123-127.
- Normann E, Naas J, Gnarpe J, et al. Demonstration of Chlamydia pneumoniae in cardiovascular tissues from children with Kawasaki disease. Pediatr Infect Dis J. 1999; 18: 72-73.
- Carter RF, Haynes ME, Morton J. Rickettsia-like bodies and splenitis in Kawasaki disease. Lancet. 1976; 2: 1254-1255.
- Weir WR, Bouchet VA, Mitford E, et al. Kawasaki disease in European adult associated with serological response to Coxiella burnetii. Lancet. 1985; 2: 504.
- Keren G, Wolman M. Can Pseudomonas infection in experimental animals mimic Kawasaki disease. J Infect dis. 1984; 9: 22-29.
- Muso E, Fujiwara H, Yoshida H, et al. Epstein-Barr virus genome-positive tubulointerstitial nephritis associated with Kawasaki disease-like coronary aneurysms. Clin Nephrol. 1993; 40: 7-15.
- Burns JC, Geha RS, Schneeberger EE, et al. Polymerase activity in lymphocyte culture supernatants from patients with Kawasaki disease. Nature. 1986; 323: 814-816.
- Lin CY, Chen IC, Cheng TI, et al. Virus-like particles with reverse transcriptase activity associated with Kawasaki disease. J Med Virol. 1992; 38: 175-182.
- Okano M, Thiele GM, Sakiyama Y, et al. Adenovirus infection in patients with Kawasaki disease. J Med Virol. 1990; 32: 53-57.
- Schulz TF, Hoad JG, Whitby D, et al. A measles virus isolate from a child with Kawasaki disease: sequence comparison with contemporaneous isolates from ’classical’ cases. J Gen Virol. 1992; 73: 1581-1586.
- Murata H. Experimental Candida- induced arteritis in mice: relation to arteritis in the mucocutaneous lymph node syndrome. Microbiol Immunol. 1970; 23: 825-831.
- Fang Y, Aravamudan VM, Sridharan GK, et al. Kawasaki like illness due to COVID-19: a review of the literature. J Infect Dev Ctries. 2021; 15: 630-638.
- Banday AZ, Arul A, Vignesh P, et al. Kawasaki disease and influenza-new lessons from old associations. Clin Rheumatol. 2021; 40: 2991-2999.
- Nakamura Y, Yashiro M, Uehara R, et al. Epidemiologic features of Kawasaki disease in Japan: results from the nationwide survey in 2007–2008. J Epidemiol. 2010; 20: 302-307.
- Rowley AH, Baker SC, Shulman ST, et al. Cytoplasmic inclusion bodies are detected by synthetic antibody in ciliated bronchial epithelium during acute Kawasaki disease. J Infect Dis. 2005; 192: 1757-1766.
- Esper F, Shapiro ED, Weibel C, et al. Association between a novel human coronavirus and Kawasaki disease. J Infect Dis. 2005; 191: 499-502.
- Sawa F. Circulating immune complexes in MCLS. Acta Paed Jap. 1979; 83: 493-498.
- Weindling AM. Levinsky RJ, Marshall WC, et al. Circulating immune complexes in mucocutaneous lymph-node syndrome (Kawasaki disease). Arch Dis Child. 1979; 54: 241-242.
- Eluthesen K, Marchette N, Melish M, et al. Circulating immunecomplexes in Kawasaki’s disease: detection of C1q binding assay. Presented at 21st inter science conference on Antimicrobial Agents and chemotherapy. November 4 to 6. 1981.
- Furuse A. Matsuda I. Circulating immune complex in the mucocutaneous lymph node syndrome. Eur J Pediatr. 1983; 141: 50-51.
- Yanase Y, Kawasaki T, Yoshinoya S, et al. A study of immune complexes in Kawasaki disease. Arerugi (Jpn J Allergol). 1984; 33: 59-65.
- Miyata K, Kawakami K, Onimaru T, et al. Circulating immune complexes and granulocytes chemotaxis in Kawasaki disease. Jpn Circ J. 1984; 48: 1350-1353.
- Takiguchi M, Tamura T, Goto M, et al. Immunological studies on Kawasaki disease. I. Appearance of Hanganutziu-Deicher antibodies. Clin Exp Immunol. 1984; 56: 345-352.
- Mason WH, Jordan SC, Sakai R, et al. Circulating immune complexes in Kawasaki syndrome. Pediatr Infect Dis. 1985; 4: 48-51.
- Ono S, Onimaru T, Kawakami K, et al. Impaired granulocyte chemotaxis and increased circulating immune complexes in Kawasaki disease. J Pediatr. 1985; 106: 567-570.
- Levin M, Holland PC, Nokes TJC, et al. Platelet immune complex interaction in pathogenesis of Kawasaki Disease and childhood polyarteritis. BMJ. 1985; 290: 1456-1460.
- Pachman LM, Herold BC, Davis AT, et al. Immune complexes in Kawasaki syndrome: review. Prog Clin Biol Res. 1987; 250: 193-207.
- Levin M, Holland PC, Novelli V. Platelet immune complex interaction in the pathogenesis of Kawasaki disease. Prog Clin Biol Res. 1987; 250: 227-237.
- Lin CY, Hwang B. Serial immunologic studies in patients with mucocutaneous lymph node syndrome (Kawasaki disease). Ann Allergy. 1987; 59: 291-297.
- Fujimoto T, Kato H, Inoue O, et al. Immune complex study of biopsy specimens from Kawasaki disease patients. Prog Clin Biol Res. 1987; 250: 209-221.
- Ohshio G, Furukawa F, Khine M, et al. High levels of IgA- containing circulating immune complex and secretory IgA in Kawasaki disease. Microbiol Immunol. 1987; 31: 891-898.
- Salcedo JR, Greenberg L, Kapur S. Renal biopsy of mucocutaneous lymph node syndrome (Kawasaki disease). Clin Nephrol. 1988; 29: 47-51.
- Salo E, Kekomaki R, Pelkonen P, et al. Kawasaki disease: monitoring of circulating immune complexes. Euro J Pediatr. 1988; 147: 377-380.
- Salo E, Pelkonen P, Kekomaki R, et al. Kawasaki disease: circulating immune complexes monitored during the disease. Prog Clin Biol Res. 1987; 250: 563.
- Li CR, Yang XQ, Shen J, et al. Immunoglobulin G subclasses in serum and circulating immune complexes in patients with Kawasaki syndrome. Pediatr Infect Dis. 1990; 9: 544-547.
- Koike R. The effect of immunoglobulin on immune complexes in patients with Kawasaki disease. Acta Paediatr. 1991; 33: 300-309.
- Philip S, Lee WC, Liu SK, et al. A swine model of horse serum- induced coronary vasculitis: an implication for Kawasaki disease. Pediatr Res. 2004; 55: 211-219.
- Philip S, Lee WC, Wu MH, et al. Histopathological evaluation of horse serum-induced immune complex vasculitis in swine: implication to coronary artery lesions in Kawasaki disease. Pediatr Neonatol. 2014; 55: 297-305.
- Knicker WT, Cochrane CG. The localization of circulating immune complex in experimental serum sickness. J Exp Med. 1968; 127: 119-135.
- Fossard C, Thompson RA. Mucocutaneous lymph-node syndrome (Kawasaki disease): probable soluble complex disorder. Br Med J. 1977; 880-883.
- Cochrane CG. Mechanisms involved in the deposition of immune complex in tissues. J Exp Med. 1971; 134: 755-758.
- Onouchi Z, Ikuta K, Nagamatsu K, et al. Coronary artery aneurysms develop in weanling rabbits with serum sickness but not in mature rabbits: an experimental model for Kawasaki disease in humans. J Vasc Dis. 1995; 46: 679-686.
- Suowen Xu, Chen M, Weng J. Letter to editor. COVID-19 and Kawasaki disease in children. Pharamacological research. 2020: 159: 104951.
- La Torre F, Leonardi L, Giardino G, et.al Immunological basis of virus-host interaction in COVID-19. Immunology Commission of the Italian Society of Pediatric Allergy, Immunology (SIAIP). Pediatr Allergy Immunol. 2020; 26: 75-78.
- Mehta P, Mcauley DF, Brown M, et al. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet. 2020; 395: 1033-1034.
- Hamideh Amirfakhryan. Kawasaki-like disease in children with COVID-19: A hypothesis. Med Hypotheses. 2020; 143: 110-117.
- Philip S, Lee WC, Cherian KM, et al. Role of Antioxidants in Horse Serum-mediated Vasculitis in Swine: Potential Relevance to Early Treatment in Mitigation of Coronary Arteritis in Kawasaki Disease. Pediatr Neonatol. 2017; 58: 328-337.
- Menikou S, Langford PR, Levin M. Kawasaki Disease: The Role of Immune Complexes Revisited. Front Immunol. 2019; 10: 1156.
- García-Pavón S, Yamazaki-Nakashimada MA, Báez M, et al. Kawasaki Disease Complicated with Macrophage Activation Syndrome: A Systematic Review. J Pediatr Hematol Oncol. 2017; 39: 445-451.
- Yeung RS. Pathogenesis and treatment of Kawasaki’s disease.Curr Opin Rheumatol. 2005; 17: 617-623.
- Ueno K, Nomura Y, Morita Y, et al. Circulating Platelet- Neutrophil Aggregates Play a Significant Role in Kawasaki Disease. Circ J. 2015; 79: 1349-1356.
- Albensi BC. What Is Nuclear Factor Kappa B (NF-κB) Doing in and to the Mitochondrion? Review article. Front Cell Dev Biol. 2019.
- Lawrence T. The Nuclear Factor NF-kB Pathway in Inflammation. Cite this article as Cold Spring Harb Perspect Biol. 2009; 1: a001651.
- Tandon VR, Verma S, Singh JB, et al. Role of antioxidants and oxidative stress in cardiovascular diseases. JK Sci. 2005; 7: 61-63.
- McCall TB, Boughton-Smith NK, Palmer RM, et al. Synthesis of nitric oxide from L-arginine by neutrophils. Release and interaction with superoxide anion. Biochem J. 1989; 261: 293-296.
- Mulligan MS, Moncada S, Ward PA. Protective effects of inhibitors of nitric oxide synthase in immune complex-induced vasculitis. Br J Pharmacol. 1992; 107: 1159-1162.